
Structure-Based Drug Design
The main goal of structure-based design is to utilize the information about target proteins, DNA, RNA, and other recently discovered biological molecules. The title of “receptor-based” design appears historically from the time when scientists thought that only receptor molecules can be a target for pharmaceutical intervention. We will use farther the term target-based design.
Protein targets: Solved crystal structures vs Predicted structures
The majority of structure-based design is done on the basis of proteins. One needs a structure of protein to start such a project. There are around two million proteins in human and estimated number of proteins in all species most probably is more than 10 mln. The protein data bank contains around 70,000 proteins structures solved by X-ray crystallography or NMR (and increased in increments of around 7–8 thousands structures per year). It means that in near 100 or more years the number of unsolved structures will still be overwhelming.
Types of design based on structures
-
Target-Based Drug Design
-
Ligand-Based Drug Design
-
Interface Drug-Design
Target-Based Drug Design
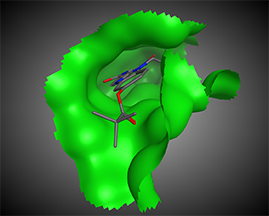
Target-based, also called receptor-based or protein-based, drug design relies on knowledge of
3D structure of the target molecule obtained through crystallography or NMR-spectroscopy methods or through homology models—predicted structures.
The
principles of target based drug design is the same whether we use a solved crystal structure or predicted structure. The difference is that the predicted structure can be somewhat different from the
real protein structure. Here the expertise of scientists who is making prediction of structure or modeling the protein hard to overestimate. A lot of failures in biotechnology had occurred because
bioinformatics specialists often overestimated the trustworthily of such predictions. Only the most experienced experts can use prediction effectively. Our company experts have extensive expertise in
protein structure prediction
Not always the reliable prediction can be done for entire protein sequences; sometimes we can trust our prediction for a small domain of a protein, but in many cases of drug design it is all we need.
The bottom line: our structure predictions are sufficiently accurate and can be used in the further structure-based drug design.
We have a protein structure: What’s the next step?
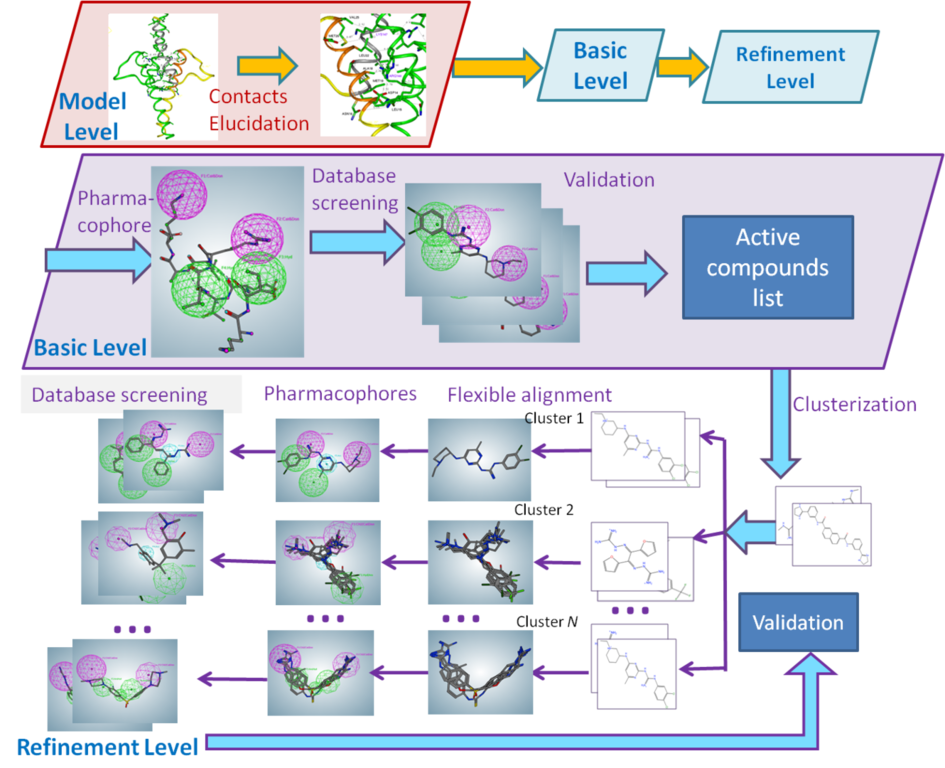
The next step is the analysis of possible points of intervention, i.e., active site identification: compound-binding pockets, dimerization interface, protein–protein-binding interfaces, protein–DNA-binding interfaces, etc.
Such an analysis will elucidate the possible centers that would be used for further pharmacophore hypothesis design.
By definition “A pharmacophore is an abstract description of molecular features, which are necessary for molecular recognition of a ligand by a biological macromolecule.”
By the IUPAC a pharmacophore is "an ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response."
A pharmacophore model explains how structurally diverse ligands can bind to a common receptor site.
After a pharmacophore is established, the set of compounds would be found using different 3D compound libraries for virtual screening. Various compounds may be found. Compounds with best activities may be used as prototypes.
Ligand-Based Drug Design
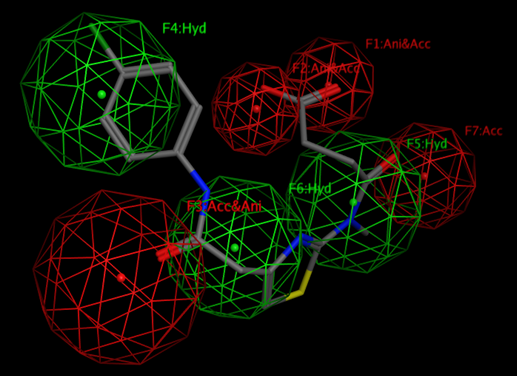
When despite all efforts we are not able to obtain a RELIABLE protein structure for examination. We highlighted the word “Reliable” because in so many cases the “enthusiastic” modelers can make a model of any protein that they get for examination. Here the severe skepticism need to be introduced and we would use only the models that we can reliably trust.
So, no target structure. In such case we need at least one or better some number of compounds that show activity vs the target (protein, or other biomolecule).
The first step in pharmacophore hypothesis elucidation in the case of several active compounds will be clusterization of their 3D structures. It is possible that some of compounds bind completely or partially different binding sites and their impact on the target biomolecule. Clusterization helps to do a preliminary separation of possible pharmacophore templates.
The next step is generation of pharmacophore hypothesis using the consensus features (centers) of compounds for each cluster. Created pharmacophore hypotheses are used for virtual screening of compound databases. BIANA has the databases including around 2 mln commercially available compounds from various countries of the world.
The hits from the compound databases are used for validation—experimental testing for activity vs the studied target biomolecule. The compounds that show activity are used for the pharmacophore refinement or for Structure-Activity Relationship (SAR) Analysis. The results of SAR are used for the second round of pharmacophore hypothesis refinement.
Interface Drug Design

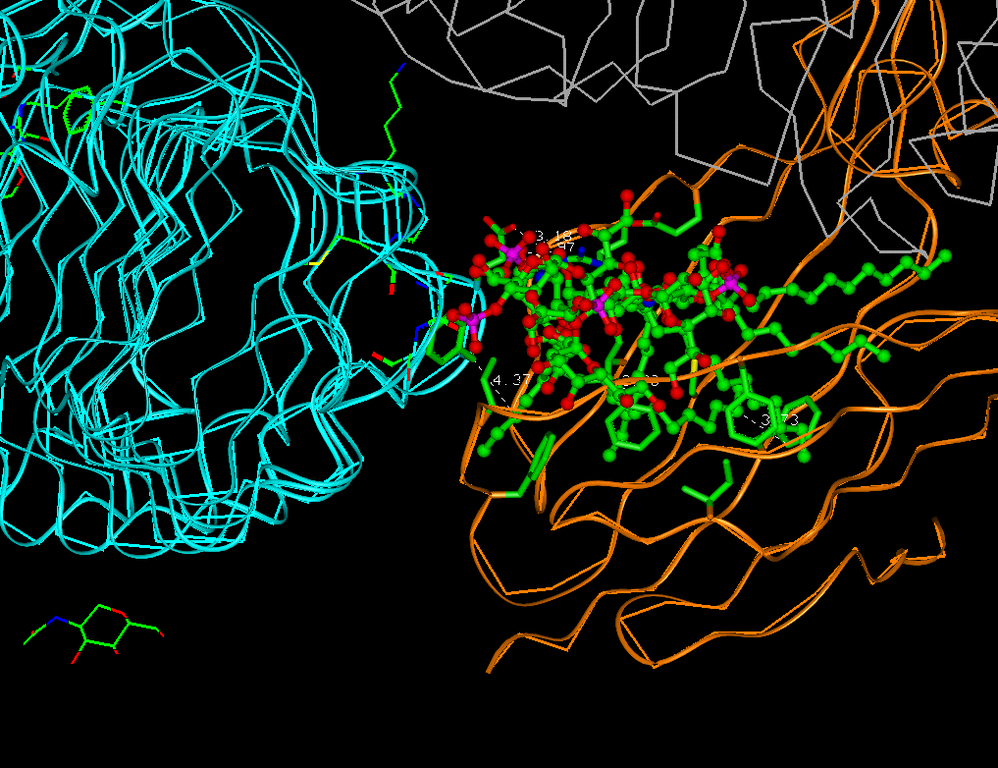
Often proteins develop dimers or trimers with the same protein helix or with other proteins and DNA. The interruption of dimerization may lead to a disease cure. The majority of the therapeutics that have currently been on the market are biomolecules, such as antisense antibodies and peptide therapies. The many disadvantages, of such therapeutics are high cost and often lack of oral bioavailability; as well as often problems with the brain-blood barrier. Small-molecules are preferable.
The main difficulty in interface drug design is to obtain a dimer or trimer structure and analyze the possible interaction sites. The protein–protein interface often is characterized by long flat surfaces, which do not have clearly defined pockets.
In general, interface drug design is similar to target-based drug design, but often a generated pharmacophore model is too large to find a good binding compound of acceptable size. For this case, we developed a method of daughter pharmacophores, because even a small molecule can interrupt the interface.
After virtual screening of compound databases, we analyze the results with theory of sets, and pick up compounds with higher probability for validation.
Additional Images
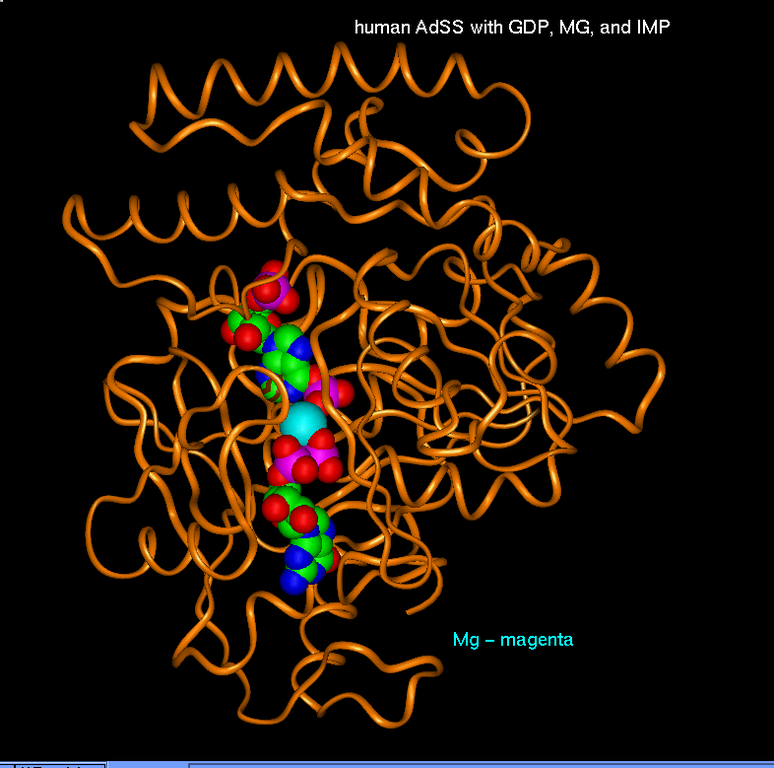
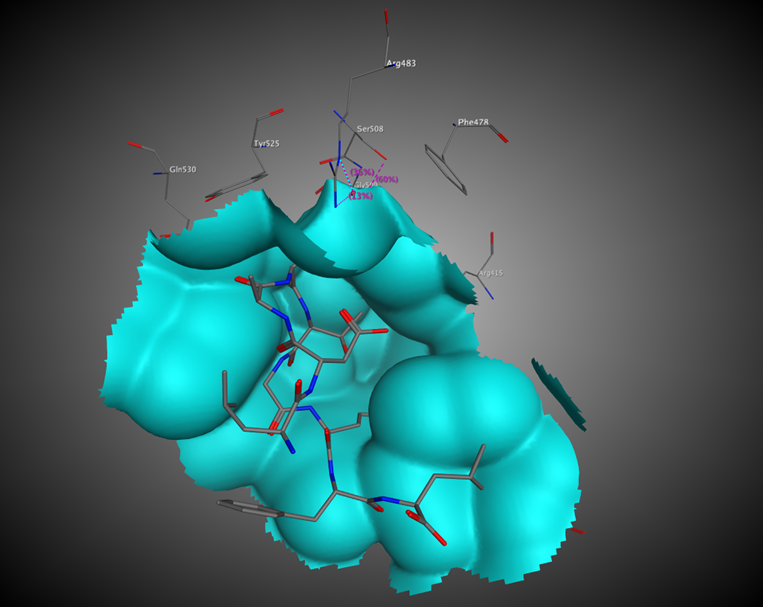

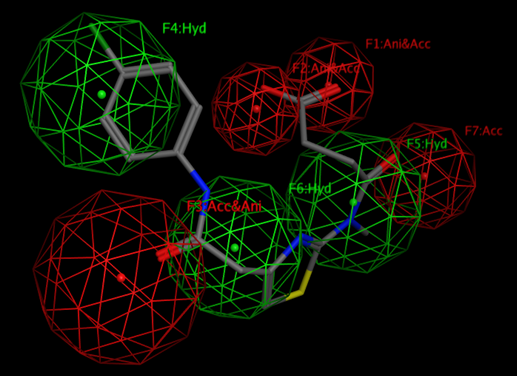
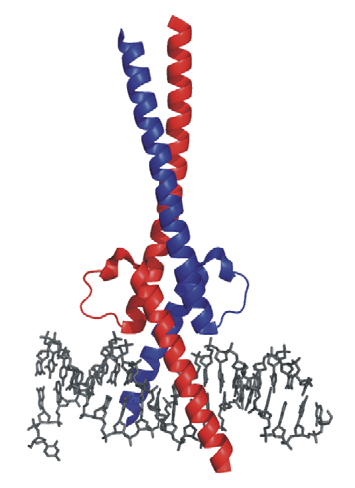
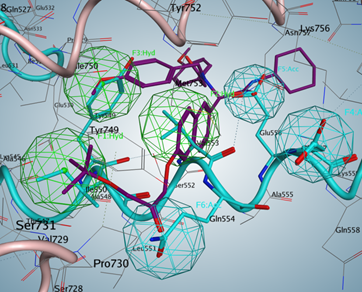
Questions?
BIANA
P.O.Box 2525
La Jolla, CA 92038
Phone: 1-858-457-0595
Fax: 1-858-581-9073
E-mail: it@biana.info
Or use our contact form.
Tip of the Week
Bring to us your project!
Lift up your reasearch on new level - make a theoretical analysis before experiments.